
Translate this page into:
A case of Peutz-Jeghers syndrome presenting as multiple intussusception of small bowel – A rare surgical emergency
*Corresponding author: Hanim Mohammed Ridwan, Department of General Surgery, Bengaluru Medical College and Research Institute, Bengaluru, Karnataka, India. hanim0705@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Srinivas NM, Pasha MM, Ridwan HM. A case of Peutz-Jeghers syndrome presenting as multiple intussusception of small bowel – A rare surgical emergency. Karnataka Med J. 2024;47:25-8. doi: 10.25259/KMJ_9_2023
Abstract
Peutz-Jeghers syndrome (PJS) is an autosomal dominant genetic disorder characterised by melanin pigment spots on the oral mucosa, lips, nasal alae, palm and soles, as well as hamartomatous polyps in the alimentary canal. Polyps are often a cause of intussusception in the affected patients. Cancers of the gastrointestinal system, uterus, and breast are common in patients with PJS. Long-term follow-up is required to prevent intussusception in children and cancer in adults. We report a classical case of PJS presenting with multiple intussusception of the small bowel in a 20-year-old male patient.
Keywords
Peutz-Jeghers syndrome
Intussusception
Polyps

INTRODUCTION
Peutz-Jeghers syndrome (PJS) is an autosomal dominant disorder caused by a mutation in the serine/threonine kinase 11/liver kinase B1 (STK11/LKB1) gene on chromosome 19, occurring with an incidence ranging from 1 in 50,000 to 200,000 people.[1] It is characterised by mucocutaneous melanotic pigmentation, gastrointestinal polyps, and increased risk of malignancy of the stomach, colon, ovary, breast, testes, small intestine, lungs and pancreas.[2]
Typically, patients experience a distinct clinical course marked by recurring episodes of bowel obstruction and bleeding induced by polyps.
CASE REPORT
A 20-year-old male presented with a 1-day history of abdominal pain, six episodes of loose stools and one episode of vomiting. He had a 2-year history of recurrent blood in stools and had undergone rectal polypectomy a year ago. Similar complaints were observed in his maternal grandfather.
Physical examination revealed melanotic pigmentation on the face, oral mucosa, lower lips and trunk [Figure 1]. The patient was febrile and tachycardic, with a distended abdomen showing diffuse tenderness, guarding, rigidity and sluggish bowel sounds.
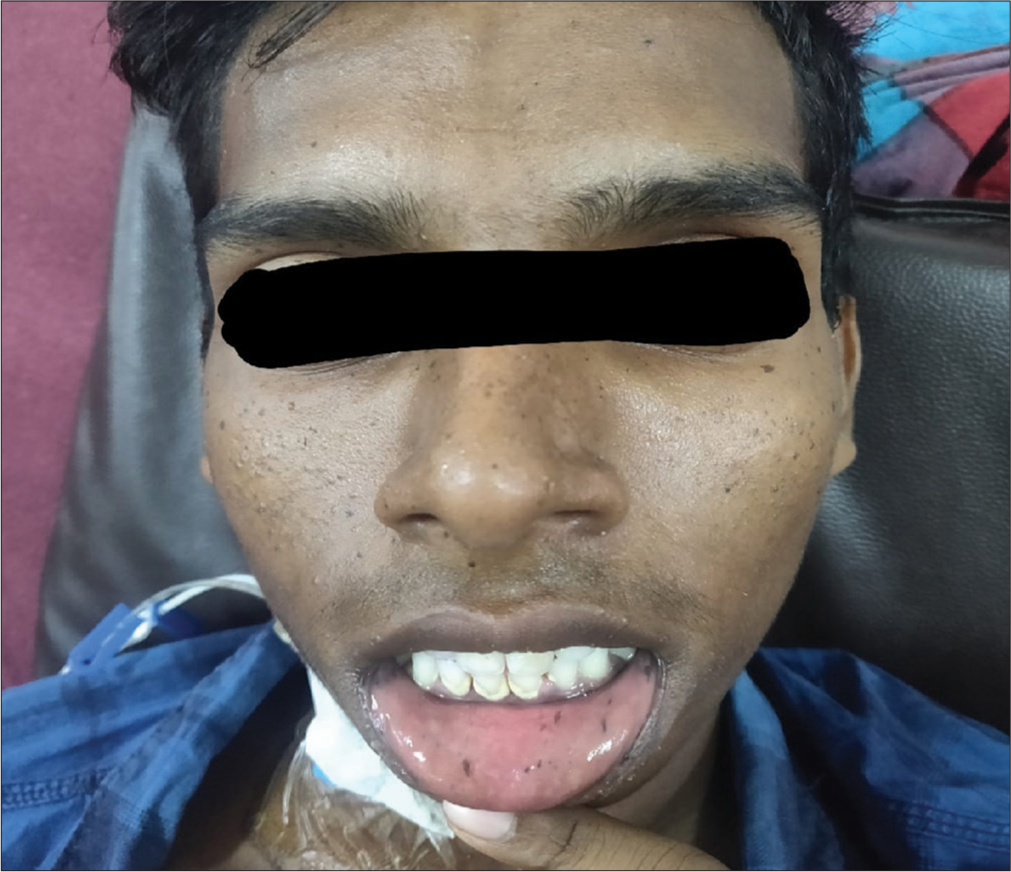
- Mucocutaneous melanotic pigmentation.
Blood and radiological tests indicated severe anaemia and features of small-bowel obstruction with intussusception.
Emergency exploratory laparotomy revealed five sites of intussusception in the small bowel, with polyps as leading points [Figures 2 and 3]. Wedge resection polypectomy was performed in three regions, and resection anastomosis was done for the remaining unhealthy bowel.
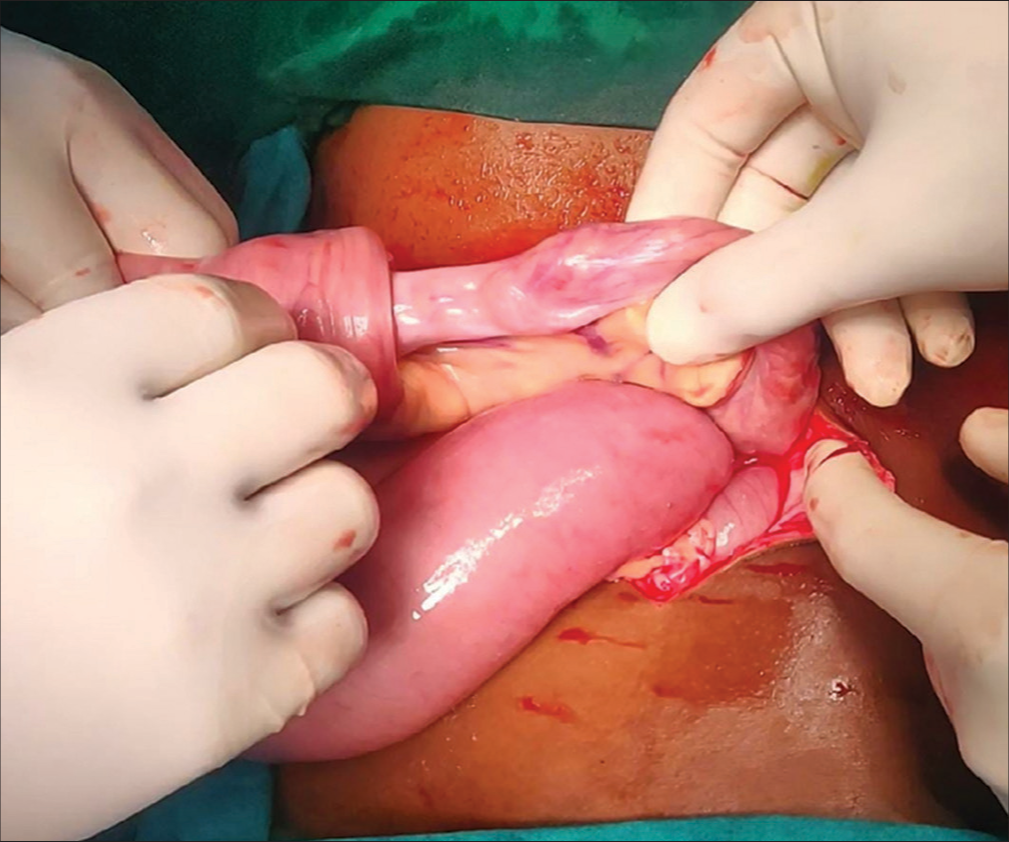
- Ileoileal intussusception.
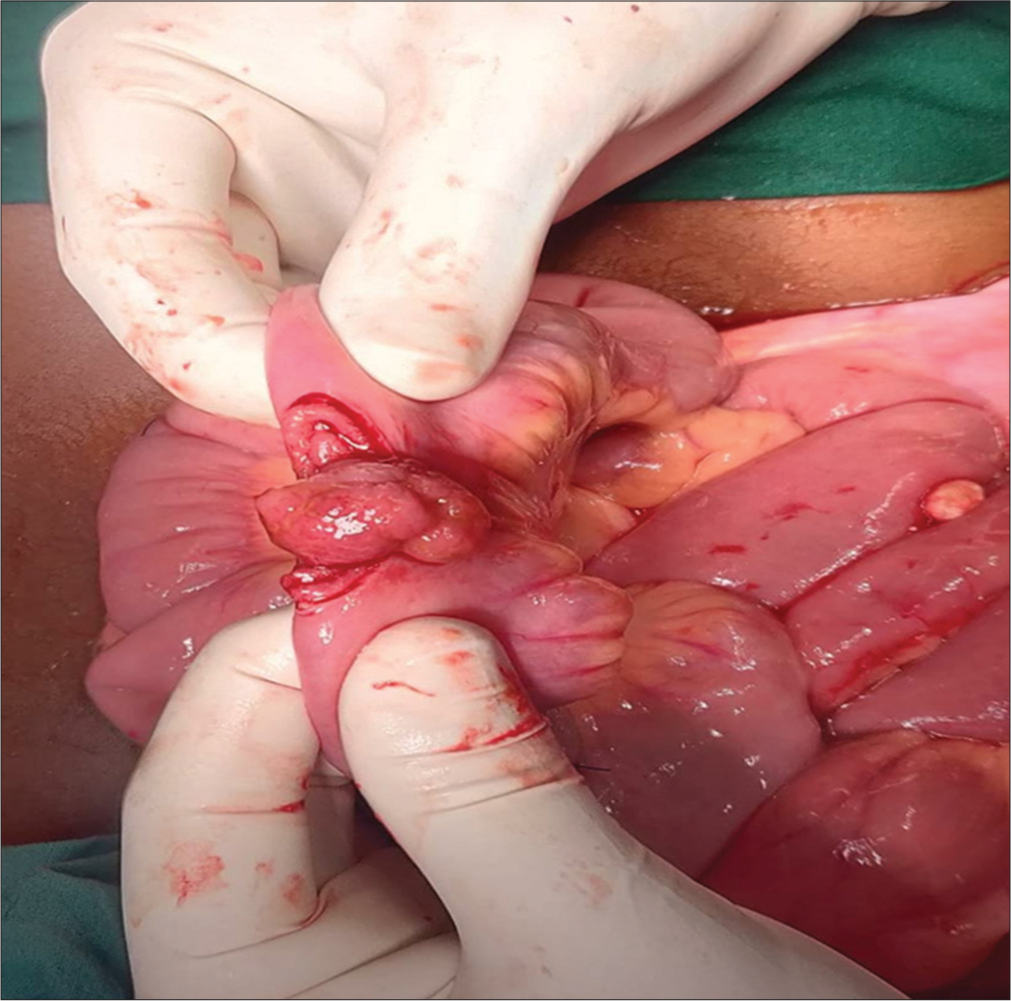
- Intussusception with polyp as leading point.
Multiple sessile and pedunculated polyps were found throughout the length of the small bowel on grossing the specimen [Figure 4]. Histopathological examination confirmed features of multiple hamartomatous polyposis [Figure 5].
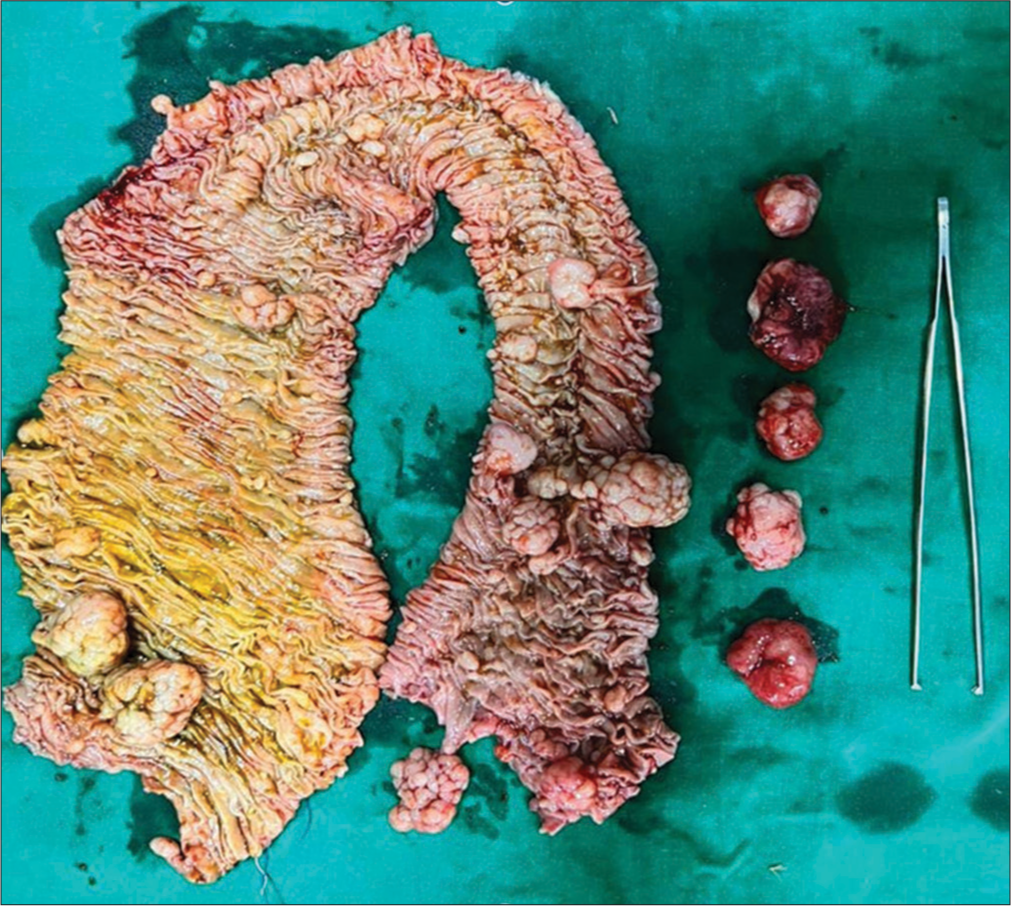
- Resected ileal loop with multiple pedunculated and sessile polyps.
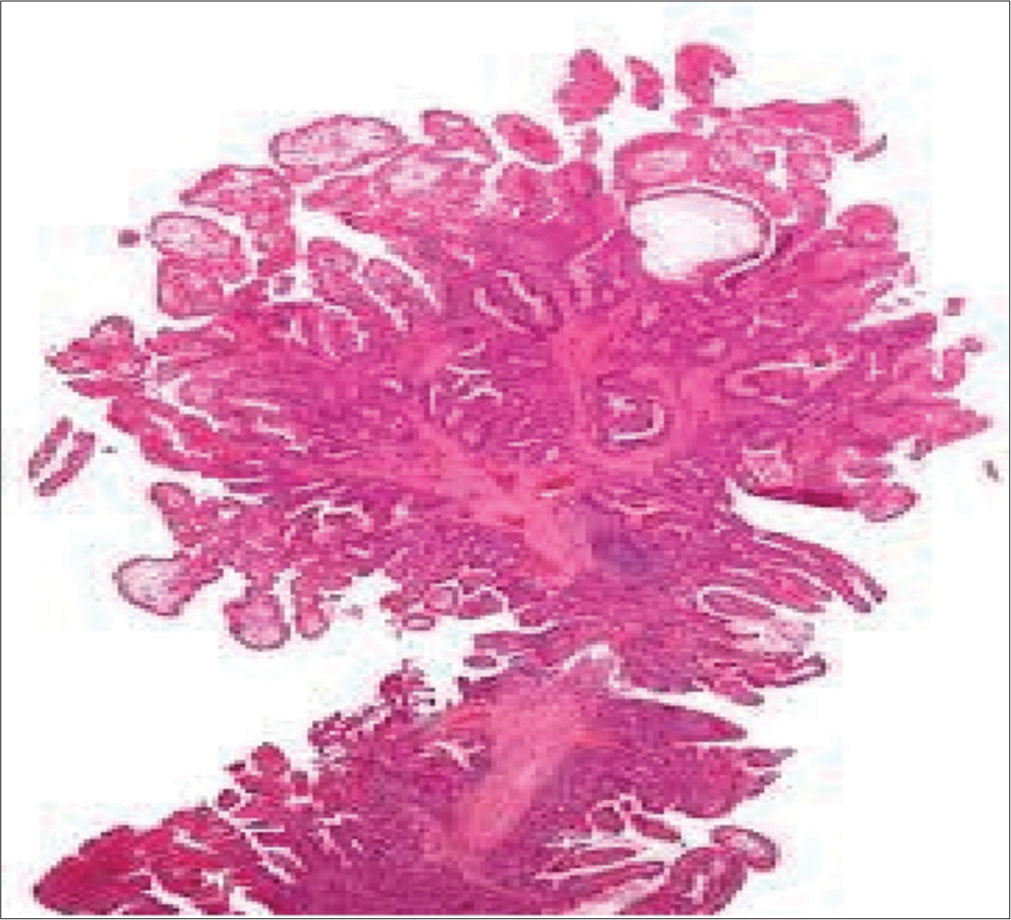
- Arborizing pattern of a hamartomatous polyp.
Postoperatively, the patient’s nutrition improved with blood transfusion and total parenteral nutrition. The patient was advised regular follow-up, colonoscopy and screening of family members.
DISCUSSION
Hutchinson was the first to describe the manifestations of PJS in 1896.[3] However, the genetic inheritance aspect was documented by Peutz in 1921.[4] The characterisation of its dominant, autosomal inheritance was later defined by Jeghers et al. in 1949.[5]
Caused by a mutation in the serine-threonine kinase (STK11/LKB1) gene on chromosome 19, this rare autosomal dominant entity manifests with mucocutaneous pigmentation, hamartomatous polyps, gastrointestinal haemorrhage, intussusception and an increased risk of malignancies. The familial form of PJS is associated with a germline mutation in the STK11/LKB1 gene. However, STK11/LKB1 mutations are infrequently observed in sporadic cases. Additional genetic abnormalities identified include overexpression of cyclooxygenase-2, mutations in the 19q13 gene, 6p11 gene and the myosin heavy chain 11 (MYH11) gene.[6]
The most distinctive clinical feature seen in almost 95% is the presence of melanin pigmentations (black-brown spots) in the lips and buccal mucosa.[6] These pigmentations can also manifest in other areas of the body, including fingers, toes, hands, feet and the mucosa of the nose, conjunctiva and rectum. They can be present at birth or appear during early infancy, and they might disappear with age, except those of the buccal mucosa, which tends to persist.
The presence of multiple hamartomatous polyps is the hallmark feature, primarily in the gastrointestinal tract, most commonly the jejunum, ileum and duodenum.[1] One-third of patients have PJS polyps in the colon and rectum as well.
Other anatomical sites that may be affected include the bronchi, gallbladder, bladder and ureter. Clinically, symptoms related to these polyps encompass bleeding, anaemia and abdominal pain resulting from intussusception, obstruction or infarction. These symptoms often manifest in childhood, with 33% of cases observed by the age of 10 years, and 50% of patients experiencing polyp-related symptoms by the age of 20 years.[6] By the age of 10 years, 30% of patients with PJS already require a laparotomy.[7]
Diagnosing PJS is based on the World Health Organisation criteria, requiring either (1) at least three hamartomatous polyps, or (2) hamartomatous polyps along with a family history of PJS, or (3) mucocutaneous pigmentation with a family history of PJS or (4) mucocutaneous pigmentation and hamartomatous polyps. Therefore, histological identification of a Peutz-Jeghers polyp plays a crucial role in confirming the syndrome. In the present case, the diagnosis was confirmed due to the hamartomatous small intestinal polyps and mucocutaneous hyperpigmentations.
Epidemiological and molecular genetic studies indicate a significant cancer risk in PJS, with a 10–18-fold excess observed in both gastrointestinal and extra-gastrointestinal malignancies.[8] Colorectal, small intestinal including the duodenum, stomach and pancreatic carcinoma represents the most common gastrointestinal malignancies reported.[9] These patients are also susceptible to various extra-intestinal tumours, including testicular Sertoli cell tumours, ovarian tumours such as sex cord tumours with annular tubules, granulosa theca cell tumours and cystadenomas. In addition, they may be at risk for breast tumours such as carcinoma breast, papilloma with squamous metaplasia, cholangiocarcinoma, pancreatic adenocarcinoma, adenoma malignum, bronchial carcinoids as well as papillomas in the bladder and pelvis.[10]
Surveillance protocols in PJS, therefore should aim to detect gastroenterological polyps that may cause obstruction, bleeding or intussusceptions as well as aim for early detection of malignancies. For individuals with PJS, a baseline colonoscopy is recommended at age 25 and should be repeated every 2–3 years thereafter. Most recommendations stress screening affected individuals with upper gastrointestinal endoscopy and small-bowel follow-through (barium study) starting at age 10 years or sooner if symptoms develop, with repeat examinations every 2 years. Video capsule endoscopy or double-balloon endoscopy may be considered to replace barium visualisation of the small bowel in PJS. Surgery is recommended for the removal of small-bowel polyps that are symptomatic or larger than 1.5 cm, particularly for polyps not accessible through endoscopy. In cases requiring laparotomy, some experts suggest intraoperative small-bowel endoscopy to ensure the removal of all identifiable polyps.[1]
Breast screening involves annual mammography for women with PJS, starting between ages 25 and 35. Annual testicular examinations should commence at age 10, and for women, annual pelvic examinations, ultrasounds and Papanicolaou tests are suggested to begin at age 20.
CONCLUSION
Patients with Peutz-Jeghers syndrome should be followed by a multidisciplinary team with the aim of the initial consultation and continued follow-up to educate the patient and family on the illness, outline a schema for continued disease surveillance, and offer genetic counselling and testing to the patient and family. Counselling and testing of asymptomatic but at-risk individuals are directed toward limiting the likelihood of patients presenting with complications, including malignancy, as well as providing preventive strategies, including cancer-surveillance measures.
Ethical approval
The research/study complied with the Helsinki Declaration of 1964.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- The concise handbook of family cancer syndromes. Mayo Familial Cancer Program. J Natl Cancer Inst. 1998;90:1039-71.
- [CrossRef] [PubMed] [Google Scholar]
- Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med. 1998;128:896-9.
- [CrossRef] [PubMed] [Google Scholar]
- A very remarkable case of familial polyposis of the mucous membrane of the intestinal tract and nasopharynx accompanied by peculiar pigmentation of the skin and mucous membrane. Ned Tijdschr Geneeskd. 1921;10:134-46.
- [Google Scholar]
- Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits. N Engl J Med. 1949;241:993-1005.
- [CrossRef] [Google Scholar]
- Peutz-Jeghers syndrome: A systematic review and recommendations for management. Gut. 2010;59:975-86.
- [CrossRef] [PubMed] [Google Scholar]
- Complications of childhood Peutz-Jeghers syndrome: Implications for pediatric screening. J Pediatr Gastroenterol Nutr. 2004;39:219-20.
- [CrossRef] [PubMed] [Google Scholar]
- WHO classification of tumours of the digestive system (4th ed). France: IARC; 2010.
- [Google Scholar]
- Peutz-Jeghers syndrome: A critical look at colonic PeutzJeghers polyps. Mod Pathol. 2013;26:1235-40.
- [CrossRef] [PubMed] [Google Scholar]
- Peutz-Jeghers syndrome In: Gastrointestinal pathology: An atlas and text (3rd ed). Philadelphia, PA: Lippincott Williams and Wilkin; 2008. p. :704-11.
- [Google Scholar]




