
Translate this page into:
Genetic inheritance in tandem: Non-syndromic autosomal recessive congenital ichthyosis with beta-thalassaemia: A rare coincidence

*Corresponding author: Arnab Ghorui, Department of Paediatrics, All India Institute of Medical Sciences, Patna, Bihar, India. arnabmck999@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Ghorui A, Kaur G, Kumar CM. Genetic inheritance in tandem: Non-syndromic autosomal recessive congenital ichthyosis with beta-thalassaemia: A rare coincidence. Karnataka Med J. 2024;47:29-34. doi: 10.25259/KMJ_12_2023
Abstract
Ichthyosis belongs to the group of Mendelian disorders of cornification. Congenital ichthyosis is inherited as an autosomal recessive trait, so it is also known as autosomal recessive congenital ichthyosis (ARCI). ARCI is classified into two types: syndromic and non-syndromic ARCI. Non-syndromic ARCIs include lamellar ichthyosis, congenital ichthyosiform erythroderma and Harlequin ichthyosis. Syndromic-ARCI is associated with multisystemic involvement, which includes Netherton syndrome, Chanarin-Dorfman disease and others. Other systemic diseases associated with ichthyosis include Gaucher disease type II and hypothyroidism. Chanarin-Dorfman and Gaucher disease-II are additionally associated with hepatosplenomegaly (HSM) and anaemia. We describe a child of congenital ichthyosis with HSM and anaemia thought to be syndromic-ARCI, but diagnosed for beta-thalassaemia (b-thal) concurrently. An infant presented with peeling skin along with absent sweating since birth and recent onset paleness of the body. Examination revealed Ichthyosis (Lamellar-variant), severe pallor and HSM, raising the possibility of Chanarin-Dorfman syndrome and Gaucher’s disease. The investigation revealed an Erlenmeyer flask deformity of the knee, but no Gaucher’s cells were found in the bone marrow. In the absence of lipid vacuoles leaden leukocytes (Jordan’s anomaly), Chanarin-Dorfman disease was also ruled out. Haemoglobin (Hb) high-performance liquid chromatography revealed β-thal major, and both parents were traits, giving us the diagnosis, which was further confirmed by the next-generation gene sequencing for clinical exomes. This report was to highlight non-syndromic ARCI involving the CYP4F22 gene variant, which is a rare finding, and the association of such ichthyosis with β-thal major was an unexpected result. Genetic counselling was provided to the parents in light of the autosomal recessive nature of both diseases. Genes of congenital ichthyosis and β-thal were unrelated, but simultaneous expressions of two autosomal-recessive diseases together are merely by chance or a new entity.
Keywords
Beta-thalassaemia
Congenital ichthyosis
Erlenmeyer flask deformity
Non-syndromic ARCI

INTRODUCTION
Ichthyosis belongs to the large and clinically and genetically heterogeneous group of Mendelian disorders of cornification (MeDOC) that can be acquired or hereditary.[1] Autosomal recessive congenital ichthyosis (ARCI) is a group of non-syndromic ichthyosis that usually presents at birth and can progress into any one of a spectrum of disorders, namely lamellar ichthyosis, nonbullous congenital erythroderma or harlequin ichthyosis.[2] Fischer identified variations in at least 1 of these genes in 78% of cases among 520 families (TGM1 in 32%, NIPAL4 in 16%, ALOX12B in 12%, CYP4F22 in 8%, ALOXE3 in 5% and ABCA12 in 5%).[3] The study by Chiramel et al. from a tertiary care centre of India reported that among the 28 patients with ARCI, 12 (42.9%) had congenital ichthyosiform erythroderma, 8 (28.6%) had lamellar ichthyosis, 5 (17.9%) had intermediate phenotype and 3 (10.7%) had bathing suit ichthyosis.[4] A similar study by Ghosh et al. from a tertiary care centre of eastern India, which included 106 cases of congenital ichthyosis, found that only 10 (9.43%) were of lamellar ichthyosis.[5] Beta-thalassaemia (b-thal) is a hereditary, autosomal recessive, chronic haemolytic anaemia caused by the reduction or complete depletion of beta-globin chains of haemoglobin (Hb). There are currently over 800 beta-globin gene (HBB gene) variants known to cause b-thal worldwide.[6] We describe a child of congenital ichthyosis with marked hepatosplenomegaly (HSM) who was diagnosed with beta-thalassaemia major concurrently. Congenital ichthyosis present with HSM can be mistaken for syndromic forms of ichthyosis like Gaucher’s disease, leading to delayed diagnosis of other associated underlying pathology, in this case, b-thal major. Despite its known implication in the development of ARCI, CYP4F22 is one of the least frequently reported ARCI genes and is mainly associated with lamellar ichthyosis, the rarest form with an incidence of less than 3 per million, making its presentation with b-thal a rare coincidence.[7,8]
CASE REPORT
An 8-month-old baby girl, born to non-consanguineous parents, presented with complaints of fever, which was insidious in onset and intermittent in nature since birth. She had a history of skin peeling and absent sweating since birth, but lacrimation was normal. There was no history of abdominal pain, crying during micturition, runny nose, redness of the eye, ear discharge, vomiting, loose stool, abnormal body movements, or drowsiness. The stool and urine frequency was normal. She was completely immunised as per the national immunisation schedule. Developmental history was up to the mark for the age. The baby was exclusively breastfed till six months of age, and complementary feeding started thereafter. Family history was unremarkable for any genetic disorder. On clinical examination, her vitals revealed tachycardia (heart rate: 126 beats/minute), tachypnoea (respiratory rate: 54 cycles/minute), normal axillary temperature (98.8°F) and normal blood pressure (76/58 mmHg, 50th–90th centile for age, gender and height). On anthropometry, her weight was 5.2 kg (<1st centile), her length was 60 cm (<1st centile), her weight for length was normal (3rd–50th centile) and her head circumference was 44 cm (50th–90th centile), so both her weight and height for age were below 1st centile with a normal weight for length suggesting underlying chronic malnutrition. General examination revealed severe pallor, with no icterus, no cyanosis, no clubbing and no lymphadenopathy. The skin was dry and scaly, which suggested ichthyosis [Figure 1]. Per-abdominal examination revealed hepatomegaly with a span of 7 cm and splenomegaly up to 2 cm below the left costal margin along its long axis [Figure 2]. A cardiovascular system examination revealed tachycardia with a pan systolic murmur over the precordium likely to be a flow murmur. Respiratory system examination revealed tachypnoea with bilateral fine basal crepitation without any other added. Neurological examination revealed all the motor, cognitive and language milestones were age-appropriate and no sign of focal neuro deficit or raised intracranial pressure. Thus, the clinical impression of ichthyosis with pallor with HSM with failure to thrive was made.
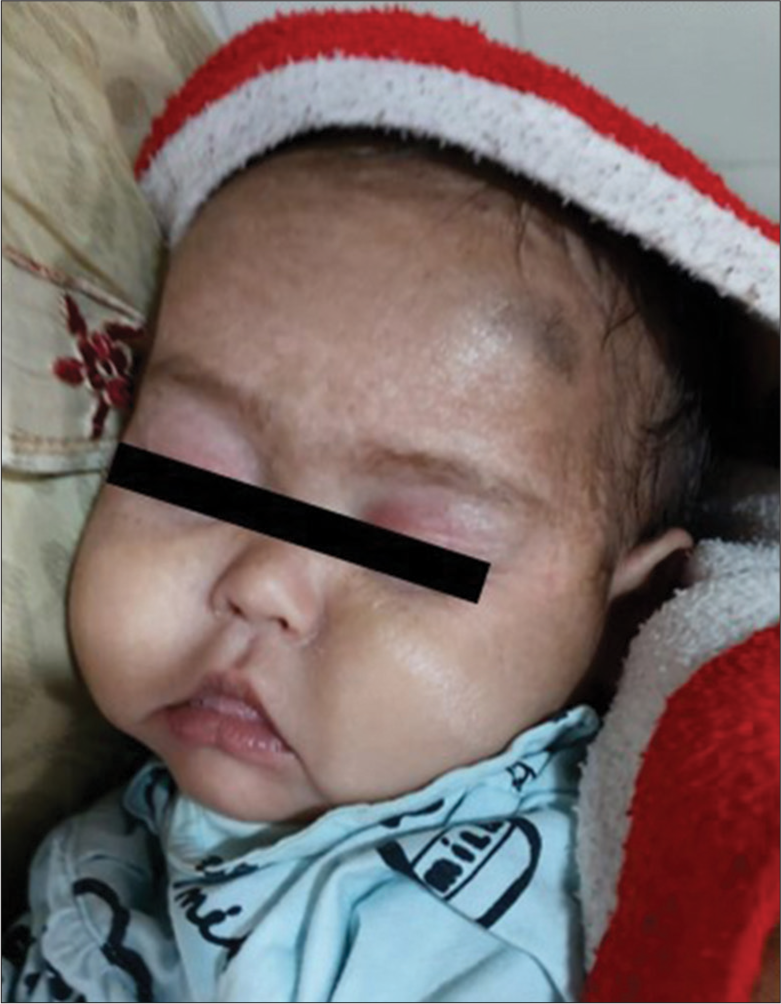
- Face with lamellar ichthyosis.
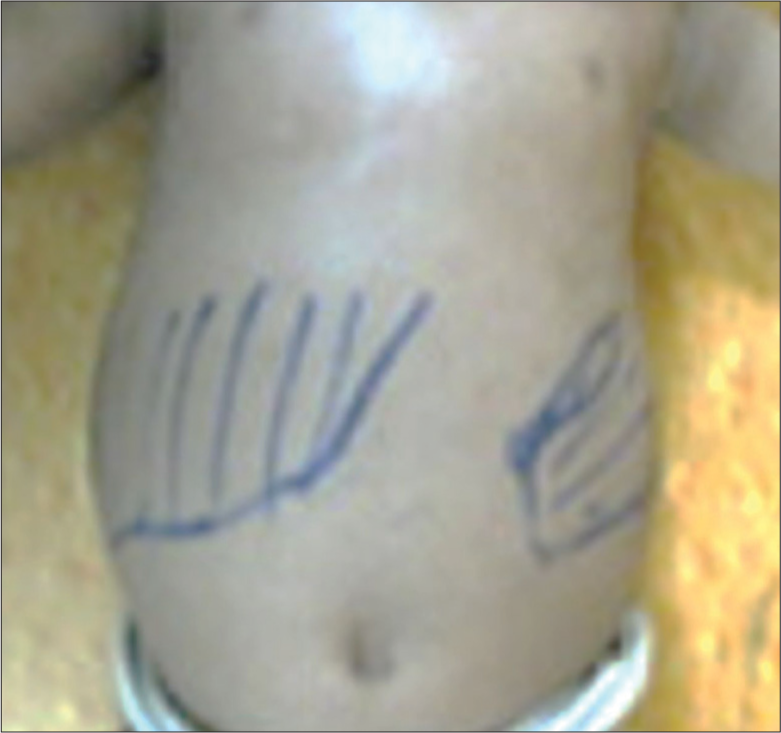
- Hepatosplenomegaly.
Laboratory investigation revealed very low Hb 3 g/dL (normal: 11–13 g/dL), normal leukocyte count 8180/mm3 (normal: 4000–11000/mm3) and low platelet count 90,000/mm3 (normal: 150,000–450,000/mm3). Peripheral blood smear examination revealed microcytic hypochromic anaemia and decreased platelet count. Her corrected reticulocyte was raised to 3% (normal: 0.5–2%), and the serum lactate dehydrogenase was also raised to 5021 U/L (normal: 170–580 U/L). The Hb high-performance liquid chromatography (HPLC) results revealed low Hb A 35.6% (normal: 94.3–98.5%), raised Hb A2 3.6% (normal: 2.2–3.5%) and raised Hb F 46.6% (normal: <2%), suggesting thalassaemia major. Then Hb HPLC was performed on both parents, which revealed that both were thalassaemia traits. Her metabolic workup revealednormal serum ammonia 30 micromol/L (normal: 11–35 micromol/L), normal lactate 1.2 mmol/L (normal: <2mmol/L), normal fasting blood sugar 96 mg/dL (normal: 70–100 mg/dL), but raised serum triglyceride 417 mg/dL (normal: <150 mg/dL). The thyroid function test was normal; thyroid-stimulating hormone 3.6 microIU/L (normal: 0.5–4.5 microIU/L) and free T4 12.63 pmol/L (normal: 9–25.7 pmol/L). The hepatic and renal function tests were normal; total bilirubin 0.6 mg/dL, aspartate aminotransferase 34.8 U/L, alanine aminotransferase 18.6 U/L, total protein 6.5 g/dL, albumin 3.6 g/dL, urea 34.5 mg/dL and creatinine 0.45 mg/dL. The serum levels of sodium and potassium were within normal limits (134 meq/L and 4.5 meq/L, respectively). Bone marrow examination revealed trilineage haematopoiesis and no storage cells. The next-generation sequencing (NGS) for clinical exomes test revealed a homozygous variant in exon 6 of CYP4F22 gene and a homozygous variant in the intron 1 of the HBB gene.
Differential diagnosis
At first glance, the absence of sweating and cracked skin pointed to ectodermal dysplasia, but the absence of defects in other ectodermal structures, such as dystrophic nails and enamel defects, ruled it out [Table 1].
| Differential diagnosis of ichthyosis | Associated feature |
|---|---|
| 1. Ectodermal dysplasia | Dystrophic nails; defective enamel |
| 2. IFAP syndrome | Alopecia |
| 3. X-linked ichthyosis | Female carriers have corneal opacities |
| 4. Congenital ichthyosiform erythroderma | Erythroderma |
| 5. Refsum’s disease | Retinitis pigmentosa |
| 6. Sjögren-Larrson syndrome | Seizures |
| 7. Epidermolytic hyperkeratosis | Characteristic pungent odour |
| 8. Netherton’s syndrome | Atopy |
| 9. Hypothyroidism | Constipation, cold intolerance |
| Differential diagnosis of ichthyosis with anaemia with HSM | Associated feature |
| 1. Gaucher’s disease | Gaucher’s cell in bone marrow biopsy |
| 2. Chanarin-Dorfman syndrome | Jordan’s anomaly |
HSM: Hepatosplenomegaly, IFAP: Ichthyosis follicularis, atrichia and photophobia
The IFAP syndrome (Ichthyosis Follicularis, Atrichia and Photophobia) was also ruled out in the absence of alopecia. The absence of comma-shaped corneal opacities on the posterior capsule eliminated the possibility of X-linked ichthyosis. Netherton’s syndrome was ruled out by the absence of the characteristic bamboo hairs. The presence of Ichthyosis with anaemia and HSM in infancy has been suspected to be an inborn error of metabolism (IEM). For the IEM workup, fasting blood sugar, arterial blood gas, serum ammonia, lipid profile, serum creatine phosphokinase, 2D echocardiography and ophthalmological evaluation were performed. The results showed hypertriglyceridemia; the rest of the parameters were unremarkable. Following these reports, the differential diagnosis narrowed down to Gaucher’s disease and neutral lipid storage disorders (NLSDs).
For Gaucher’s disease, an X-ray of the knees was performed, which revealed cortical thinning with widening of the distal end of the femur, which is characteristic of Erlenmeyer flask deformity [Figure 3]. A bone marrow biopsy was performed to confirm the diagnosis of Gaucher’s disease, but there were no Gaucher’s cells were found, so Gaucher’s disease was ruled out. For NLSD, peripheral blood examination for the presence of lipid vacuoles within leukocytes was done, which came out to be negative (absence of Jordan’s anomaly); thus, NLSD as Chanarin-Dorfman syndrome was ruled out. In this case, the history of dry, scaly skin with absent sweating since birth pointed toward the MeDOC. For further confirmation, NGS for clinical exomes was carried out, which revealed a homozygous variant in exon 6 of the CYP4F22 gene and a homozygous variant in intron 1 of HBB gene [Table 2]. The CYP4F22 gene variation is known to be involved in the pathogenesis of non-syndromic ARCIs (lamellar ichthyosis), while HBB gene variant is reported to be the culprit in the pathogenesis of thalassaemia. Thus, the constellation of clinical, haematological and genetic testing aids in the final diagnosis of non-syndromic ARCI coexisting with b-thal major.
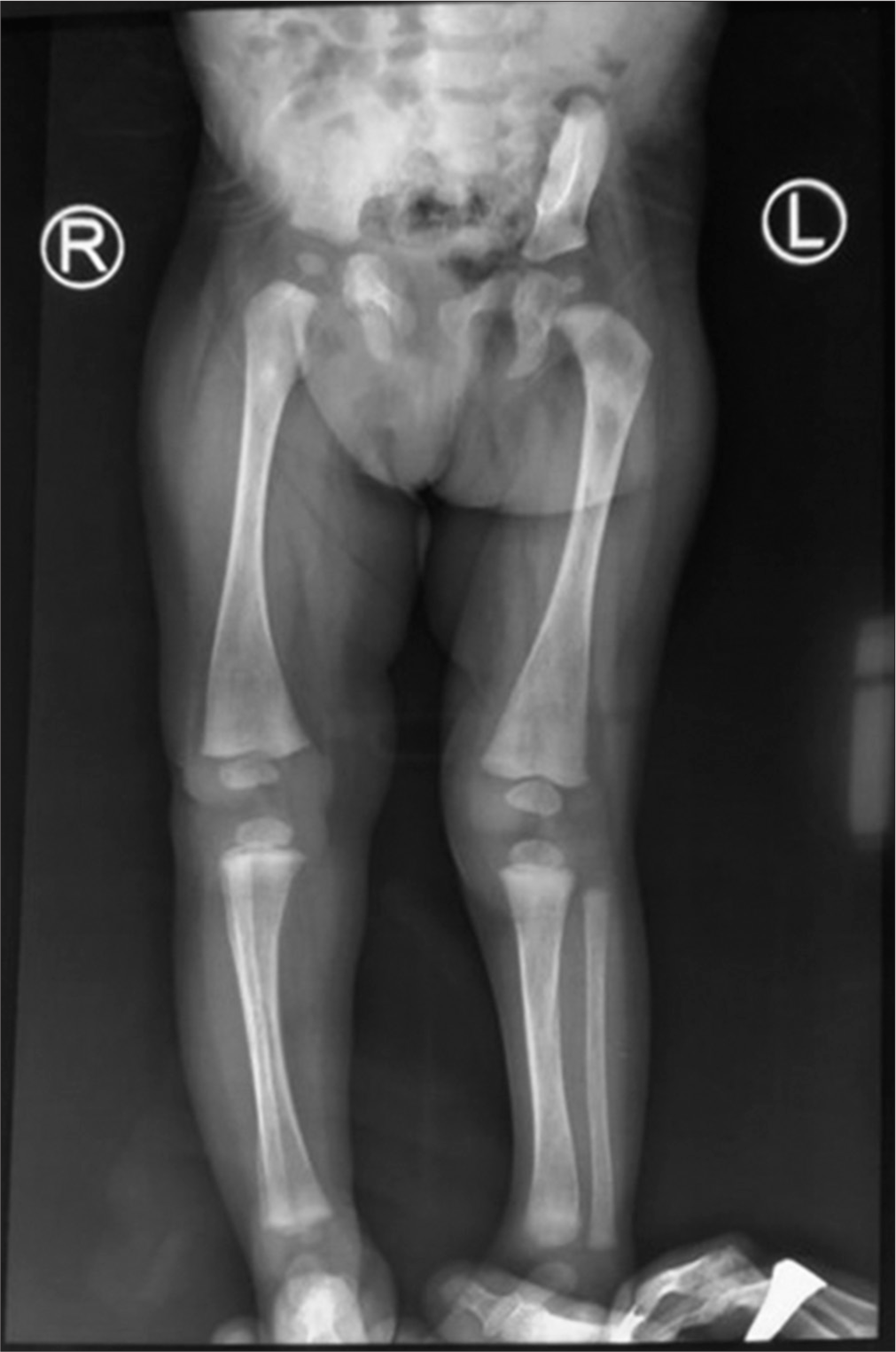
- Erlenmeyer flask deformity.
| Investigations | Results | Reference range |
|---|---|---|
| Hb | 3 g/dL | 11–13 mg/dL |
| TLC | 8180/mm3 | 4000–11000/mm3 |
| Absolute neutrophil count | 4010/mm3 | 2000–7000/mm3 |
| Platelet count | 90,000/mm3 | 150,000–450,000/mm3 |
| Peripheral blood smear | Microcytic hypochromic anaemia | ---- |
| Corrected reticulocyte | 3% | 0.5–2% |
| LDH | 5021 U/L | 170–580 U/L |
| Hb A | 35.6% | 94.3–98.5% |
| Hb A2 | 3.6% | 2.2–3.5% |
| Hb F | 46.6% | 0.0–2.0% |
| Serum ammonium | 30 µmol/L | 11–35 µmol/L |
| Serum triglyceride | 417 mg/dL | <150 mg/dL |
| CPK total | 36 U/L | 24–195 U/L |
| TSH | 3.6 µIU/L | 0.5–4.5 µIU/L |
| fT4 | 12.63 pmol/L | 9–25.7 pmol/L |
| fT3 | 3.82 pmol/L | 3.5–10 pmol/L |
| Bone marrow examination | Trilineage haematopoiesis, no storage cells | |
| NGS | CYP4F22 (+) (ENST00000269703.8), Exon 6 c. 466C >T (p.Arg156Cys), Homozygous Congenital ichthyosis-5, Autosomal recessive, Likely pathogenic |
|
| HBB (−) (ENST00000647020), Intron 1 c. 92+5G >C (5’ Splice site), Homozygous Beta-thalassaemia, Autosomal recessive, Likely pathogenic |
||
NGS: Next-generation sequencing, TSH: Thyroid-stimulating hormone, fT3: Free triiodothyronine, fT4: Free thyroxine, CPK: Creatine phosphokinase, Hb: Haemoglobin, TLC: Total leucocyte count, TSH: Thyroid-stimulating hormone, LDH:Lactate dehydrogenase, CYP4F22:Cytochrome P450, family 4, subfamily F, polypeptide 22, HBB:Haemoglobin subunit beta
RESULTS
The child in our case had congenital ichthyosis along with b-thal major. To treat the child’s anaemia and maintain pretransfusion haemoglobin levels over 10 g/dL, multiple packed red blood cell (RBC) units were transfused. Following consultation with a dermatologist, the child was treated for Ichthyosis with emollient cream and topical antibiotic cream to cure skin infection. The child’s family was somewhat hesitant to follow-up with the child despite the thorough explanation and advice given at discharge regarding the need for regular follow-up and blood transfusions. Unfortunately, the child passed away two months ago, and the untreated anaemia appears to be the cause of death. Genetic counselling was provided to the parents in light of the autosomal recessive nature of both diseases, which raised the possibility that it would be passed down through subsequent generations. Genetic testing was advised for both parents to detect the carrier state, but they were unable to do so due to financial constraints. Parents were also advised to undergo prenatal testing for the culprit genes in the subsequent pregnancy, either by chorionic villi sampling in the first trimester between 10 and 12 weeks or by amniocentesis in the second trimester between 15 and 18 weeks.
DISCUSSION
The term ichthyosis originated from the Greek word ‘ichthys’ and refers to a set of scaly skin disorders. However, the term is also used to infer particular diseases, such as lamellar ichthyosis.[2] Inherited ichthyoses are genetic illnesses distinguished by generalised drying, hyperkeratosis, and scaling of the skin.[9] Conceptually, ARCI is an effective term that comprises multiple clinical phenotypes, each of which may be tough to specifically diagnose on the grounds of appearance but which share congenital onset and pattern of inheritance. Three well-defined clinical phenotypes under this group: Lamellar ichthyosis, congenital ichthyosiform erythroderma and harlequin ichthyosis.[2] Dumenigo et al. found the following two cases that strongly support our diagnosis for skin findings, namely a 2-day-old male presented to the dermatology consult service after diffuse, thick scaling was noted at birth.[7] Nails and hair were noted to be normal, and he had no ectropion or bullae. Clinically, the patient was diagnosed with congenital non-syndromic ichthyosis.[10] A 3-year-old Chinese origin born of non-consanguineous Chinese parents was ichthyotic at birth. After birth, she presented clinical aspects of generalised lamellar ichthyosis with a brown plate-like scale; her parents had no history of skin disorders.[11] ARCI is a heterogeneous group presenting at birth having 14 subtypes (OMIM ARCI 1–14).[12] Only 8% of ARCI cases are linked to a CYP4F22 variant, making it extremely rare.[13] CYP4F22 is a protein of the cytochrome-P450 family four and codes for an epidermal w-hydroxylase crucial for the synthesis of w-hydroxy acylceramides in the epidermis, which is presumed to be important for epidermal barrier function.[14] The impaired skin barrier role may lead to unstable body temperature, dehydration, and increased vulnerability to infections.[15] In a sizable cohort of 770 families, Hotz et al. investigated patients having a clinical diagnosis of ARCI. The affected people were born with ichthyosis symptoms and no signs of a systemic illness. They discovered CYP4F22 gene variants in 54 families. Variants most commonly reported in their cohort study were p.His435Tyr, p.Arg156Cys, p.Arg156His, p.Arg243His and p.Arg243Cys.[16] There is currently little evidence in the literature about genotype-phenotype correlations for ARCI with CYP4F22 variants. Patients with CYP4F22 variants have a very modest phenotype but they were unable to discover any evidence linking the type of variant and the severity of the disease. In their study, they also evaluated missense variants affecting the putative trans-membrane domain (TMD) and substrate-binding regions (SBR) to see if these variants would cause a more severe phenotype because these two regions are crucial for the functionality of the protein. Two novel missense variants, as well as the well-known missense mutants p.Arg156Cys were all found in the SBR domain. In the TMD domain, c.59dup was found to be located. However, there is no conclusive proof that having one or both variants in these domains results in a more severe phenotype. Although, it should be noted here that the p.Arg156Cys variation of the CYPF22 gene is present in the patient case reported in our investigation.[14] In the year 2017, Gruber et al. reported a case series of two siblings with ARCI who had an identical homozygous splice-site mutation in CYP4F22. This was a rare example. While the sister was born with dry skin and no collodion membrane, the first girl was born with collodion membrane and had contractures of the great joints. Both sisters displayed a generalised fine-scaling phenotype at the age of 6 months.[17] The birth of a newborn with a MeDOC often comes with little warning to the parents.[18] Whereas cases of b-thal become severely anaemic and need of transfusion before the age of two years; thus, they are more likely to seek medical attention. b-thal, an autosomal recessive chronic haemolytic anaemia, is characterised by diminished Hb production as a result of a reduction or absence of b-globin chains. In the absence of a beta-globin chain, the unbound alpha-globin chains form a tetramer and precipitate over the RBC membrane, leading to destruction inside the bone marrow and causing erythropoiesis to be ineffective. b-thal is characterised by variableness in the clinical manifestation that is discerned by variants in the b-globin gene (HBB gene). Till today, according to the human gene mutation database (®) report, over 200 variations are known in the HBB gene that is related to b-thal. Individuals with thalassaemia major have severe anaemia and HSM; they usually come to medical attention within the first 2 years of life. Thus, a disorder that begins merely as haemolytic anaemia reaches the dimension of a chronic disease with multisystem involvement. Recognition of biallelic pathogenic variants in HBB gene on molecular genetic testing may be helpful for diagnosis in at-risk individuals under age 12 months.[19]
In this case, an 8-month-old girl child, with an unremarkable family genetic history, presented with dry, scaly skin and was found to have HSM on abdominal examination. Suspicion was raised about syndromic forms of congenital ichthyosis, but the Hb HPLC report revealed b-thal major. This report aimed to highlight non-syndromic ARCI involving the CYP4F22 gene variant, which is a rare finding, and the association of such ichthyosis with b-thal major was an unexpected result. Precise examination and a broadened investigation were the key to the final diagnosis of this patient which would have been missed if every symptom was not investigated thoroughly.
CONCLUSION
The genes of congenital ichthyosis and b-thal are unrelated but simultaneous expression of two autosomal-recessive diseases together raises a question of whether this occurrence is merely by chance or a new entity.
Congenital ichthyosis, an autosomal recessive condition, was observed in conjunction with b-thal major, another autosomal recessive condition
Systemic abnormalities in congenital ichthyosis can be mistaken for syndromic forms of ichthyosis or other congenital disorders like Gaucher disease
Congenital Ichthyosis presented with a variant in one of the least frequently reported genes, that is CYP4F22.
Ethical approval
The research/study complied with the Helsinki Declaration of 1964.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Non-syndromic types of ichthyoses-an update. J Dtsch Dermatol Ges. 2014;12:109-21.
- [CrossRef] [PubMed] [Google Scholar]
- Ichthyosis: Etiology, diagnosis, and management. Am J Clin Dermatol. 2003;4:81-95.
- [CrossRef] [PubMed] [Google Scholar]
- Autosomal recessive congenital ichthyosis. J Invest Dermatol. 2009;129:1319-21.
- [CrossRef] [PubMed] [Google Scholar]
- Genotype of autosomal recessive congenital ichthyosis from a tertiary care center in India. Pediatr Dermatol. 2022;39:420-4.
- [CrossRef] [PubMed] [Google Scholar]
- Clinicoepidemiological study of congenital ichthyosis in a tertiary care center of Eastern India. Indian J Dermatol. 2017;62:606-11.
- [CrossRef] [PubMed] [Google Scholar]
- Beta thalassemia in 31,734 cases with HBB gene mutations: Pathogenic and structural analysis of the common mutations; Iran as the crossroads of the Middle East. Blood Rev. 2016;30:493-508.
- [CrossRef] [PubMed] [Google Scholar]
- CYP4F22-related autosomal recessive congenital ichthyosis: Clinical presentation. Cureus. 2022;14:e22272.
- [CrossRef] [Google Scholar]
- Novel CYP4F22 mutations associated with autosomal recessive congenital ichthyosis (ARCI). Study of the CYP4F22 c.1303C>T founder mutation. PLoS One. 2020;15:e0229025.
- [CrossRef] [PubMed] [Google Scholar]
- Quality of life and clinical characteristics of self-improving congenital ichthyosis within the disease spectrum of autosomal-recessive congenital ichthyosis. J Eur Acad Dermatol Venereol. 2022;36:582-91.
- [CrossRef] [PubMed] [Google Scholar]
- Novel compound heterozygous mutations in the CYP4F22 gene in a patient with autosomal recessive congenital ichthyosis. Clin Case Rep. 2021;9:e05082.
- [CrossRef] [PubMed] [Google Scholar]
- Inherited ichthyosis: Non-syndromic forms. J Dermatol. 2016;43:242-51.
- [CrossRef] [PubMed] [Google Scholar]
- Autosomal recessive congenital ichthyosis. Actas Dermosifiliogr. 2013;104:270-84.
- [CrossRef] [PubMed] [Google Scholar]
- Essential role of the cytochrome P450 CYP4F22 in the production of acylceramide, the key lipid for skin permeability barrier formation. Proc Natl Acad Sci U S A. 2015;112:7707-12.
- [CrossRef] [PubMed] [Google Scholar]
- Lamellar ichthyosis in a female neonate without a collodion membrane. Dermatol Online J. 2018;24:13030/qt24g7w9t8.
- [CrossRef] [Google Scholar]
- Mutation update for CYP4F22 variants associated with autosomal recessive congenital ichthyosis. Hum Mutat. 2018;39:1305-13.
- [CrossRef] [PubMed] [Google Scholar]
- Morphological alterations in two siblings with autosomal recessive congenital ichthyosis associated with CYP4F22 mutations. Br J Dermatol. 2017;176:1068-73.
- [CrossRef] [PubMed] [Google Scholar]
- Care of the newborn with ichthyosis. Dermatol Ther. 2013;26:1-15.
- [CrossRef] [PubMed] [Google Scholar]
- The spectrum of beta-thalassemia mutations in the 22 Arab countries: A systematic review. Expert Rev Hematol. 2021;14:109-22.
- [CrossRef] [PubMed] [Google Scholar]




